

公司新闻
科研成果 | 上海交大低碳学院纪亚团队在Energy Storage Materials 发表水系有机靶向液流电池电位预测研究成果
近日,公司纪亚团队在国际知名期刊Energy Storage Materials(IF=20.4)发表了题为“Potential prediction in aqueous organic redox-targeting flow batteries: DFT calculation and experimental validation”的研究论文。该文章基于密度泛函理论(DFT)和实验验证,提出了一种精确的电位预测方案来解决水系有机靶向液流电池中氧化还原介质和固相材料筛选难题,耦合分子静电势(ESP),吉布斯自由能(GFE)与HOMO-LUMO gap计算,精准预测并成功构建电位匹配的靶向体系,电池容量提升3.86倍,固相材料利用率达到70.79%。论文第一作者为低碳学院博士研究生荣思达,通讯作者是纪亚助理教授。
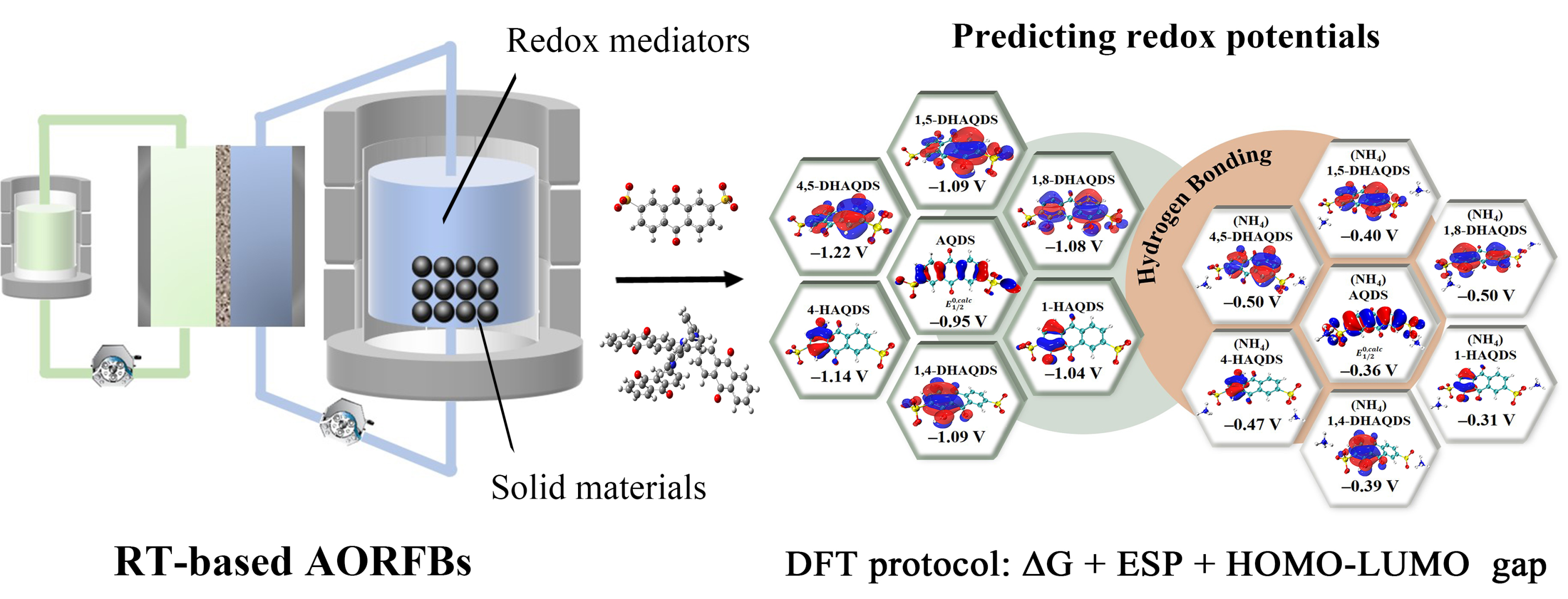
图1. 水系有机靶向液流电池中的DFT预测方案。
【研究背景】
能源安全、温室气体排放和空气污染已成为可持续发展面临的最紧迫挑战。可再生能源是应对这些挑战的有效解决方案。然而,可再生能源不可控的波动性和随机性会造成电网不稳定和严重的电力浪费。液流电池(RFB)因其能量与功率解耦、高可扩展性、设计灵活性和长周期循环等特性,成为有前景的大规模储能技术。近年来,水系有机液流电池(AORFB)脱颖而出,它利用具有可设计结构的高性价比有机分子作为 RFB中的活性物质。氧化还原靶向(RT)反应有效解决了AORFB的溶解度难题,但靶向体系面临着氧化还原介质(RM)与固相储能材料(SM)电位匹配困难的挑战,存在随机性大、成本高、耗时长等问题。因此,精准预测RM和SM的氧化还原电位的极为重要。本项目开发了一种基于密度泛函理论的预测方案,并成功进行了实验验证,对指导靶向液流电池构建具有重要意义。
【本文要点】
要点一:通过静电势与吉布斯自由能计算RM与SM的氧化还原电位
由于磺酸基团具有很强的抽电子效应,位于 1 , 8 位的羟基会被相邻的磺酸基团部分抵消其供电子效应,从而导致 DHAQDS 核心内的电子密度分布更加均匀。根据分子 ESP 极值分布,由于羟基的部分中和作用,磺酸基团氧原子上的负电荷更加集中。相反,位于 4 或 5 位的羟基离位于 2 和 6 位的磺酸基团较远,羟基无法抵消磺酸基团的夺电子效应。在 RT 反应中,理论计算显示 1,5-DHAQDS、1,4-DHAQDS 和 1,8-DHAQDS 的电位值与 PAQPy 更为匹配,表明它们有望成为基于 RT 的 AORFB 的偶联物。为了更真实地计算氧化还原电位 (RPs),在 AQDS 衍生物和 PAQPy 中加入了NH4+,以研究氢键的影响。为了确定目标分子与NH4+ 之间的确切结合位点,我们计算了 ESP,以获得精确的几何优化。高负 ESP 区域(富电子)容易吸引氢原子并形成氢键。通过三维 ESP 等值面图,可以直观地看到 ESP 达到特定阈值的区域。参与氢键的负电原子上是否存在孤对电子由电子结构决定,而电子结构共同决定了分子形成氢键的位置。
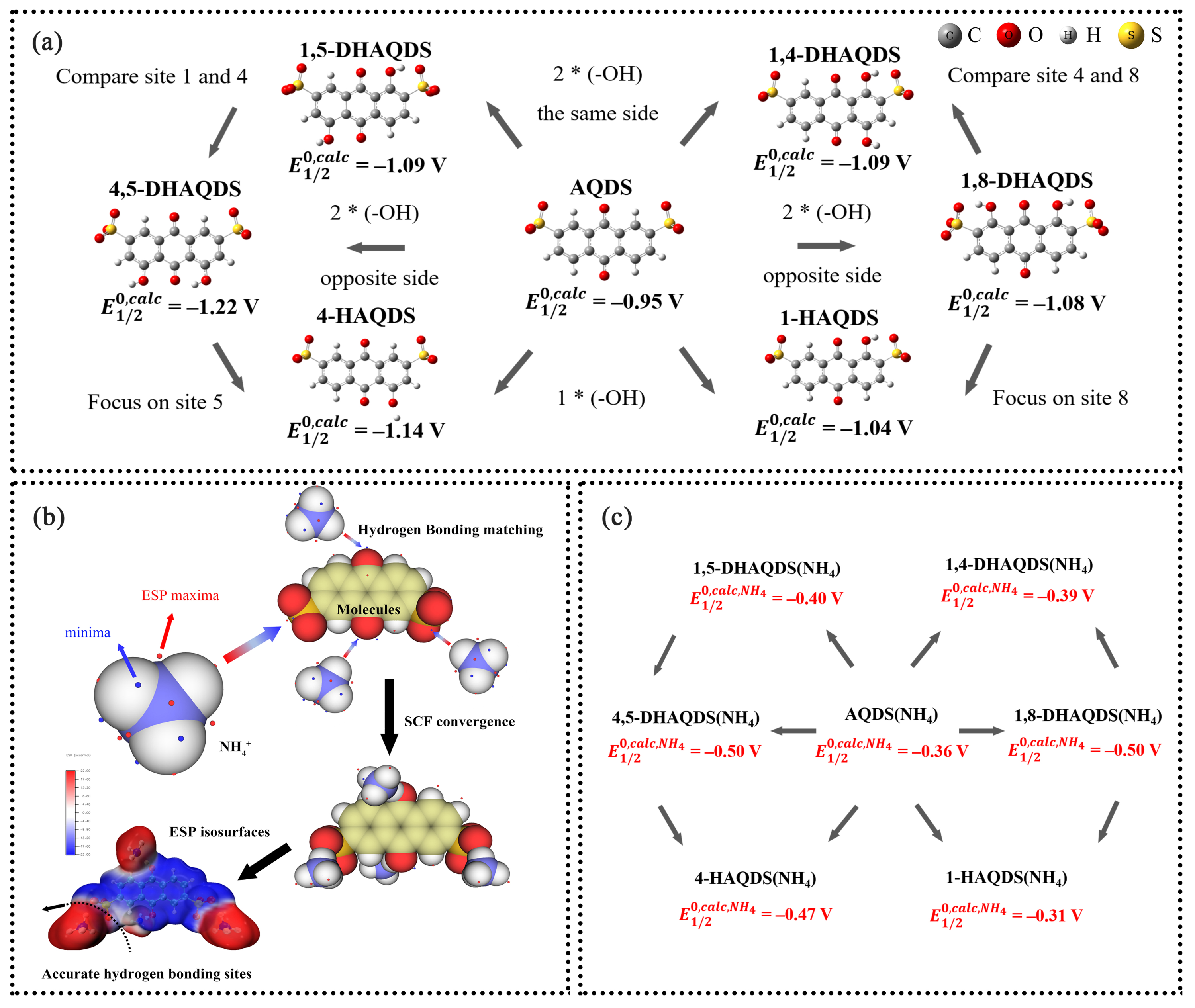
图2. 通过 DFT 计算的不含/含 NH4+ 的 AQDS 衍生物的 RPs:(a)不含NH4+的 AQDS 衍生物;(b)使用 ESP 计算氢键位点的过程;(c)含NH4+的 AQDS 衍生物。
要点二:通过氢键键长键角分析分子与环境配体相互作用
氢键通过影响电子分布、分子相互作用和氧化还原反应,在调节 RPs 方面具有重要意义。在磺酸基的同一侧,AQDS(NH4) 的 N-H 和 H...O 键长度相同,键角为 174.86°,氢原子距离共轭负电平面 2.15 Å。相反,磺酸基对侧的 DHAQDS(NH4),N-H 键长度减少到 1.12 Å,H...O 键长度增加到 1.45 Å,NH4+ 围绕羰基氧原子翻转,角度为 91.38°,氢原子距离共轭负电平面 2.47 Å。N-H...O 键的角度为 174.60°。磺酸盐基团同侧的 N-H 和 H...O 的结合模式因 -OH 取代而发生了显著变化。从不同角度看,单体羰基两侧的氢键具有明显的对称性。与 AQDS 及其衍生物不同,NH₄+ 与 sp² 杂化平面的偏差极小,没有出现分子内氢键。氢键的建立会导致电子云的重组。这一过程缩短了电子隧穿距离,降低了电子转移的 GFE 差值,从而导致 RP 正移。
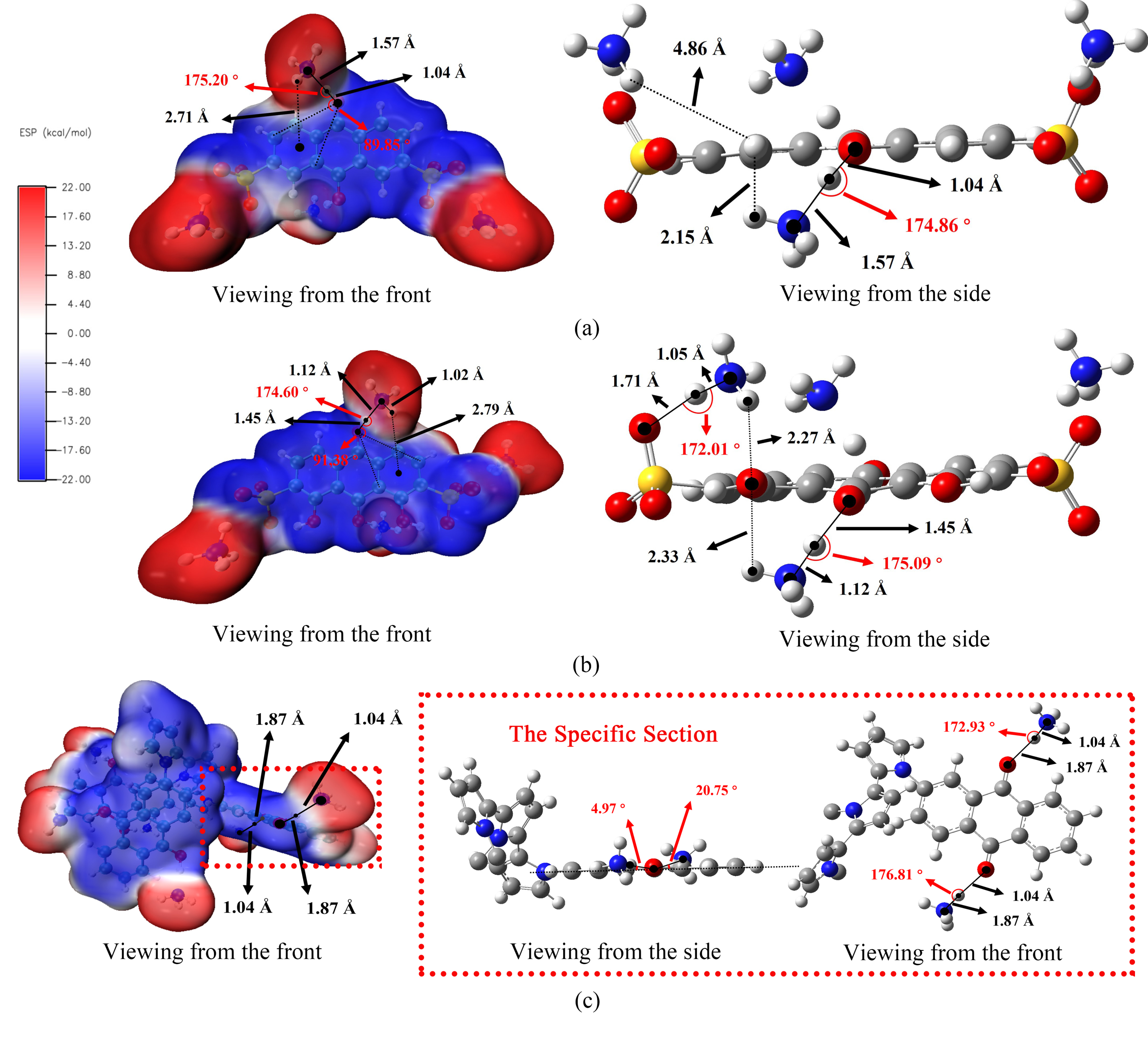
图3. 从不同的观察角度分析含有 NH4+ 的分子中氢键的长度和角度:(a) AQDS; (b) 1,8-DHAQDS 和 (c) PAQPy。
要点三:通过HOMO-LUMO gap分析氢键作用机理
根据费米级计算分子与NH₄+的 HOMO-LUMO gap,在与 NH4+ 形成氢键后,电子密度重叠明显增加。因此,由于有利的轨道对称匹配,HOMO-LUMO gap被扩大。在电子转移或氧化还原反应过程中,电子数的变化导致分子不同价态的 HOMO 和 LUMO 能级位置不同。与 NH₄+ 形成氢键后,HOMO 电子密度区域发生了显著变化,表明电子分布范围与 HOMO 能级之间存在反相关性。有机分子的 HOMO 轨道与 NH₄+一起充当供体,提供电子密度,与带负电荷的区域相互作用。NH₄+的 LUMO 与 HOMO 有类似的规律,导致 HOMO 和 LUMO 的电子密度区域分别重叠,sp2杂化的共轭面导致氢键强度更强。
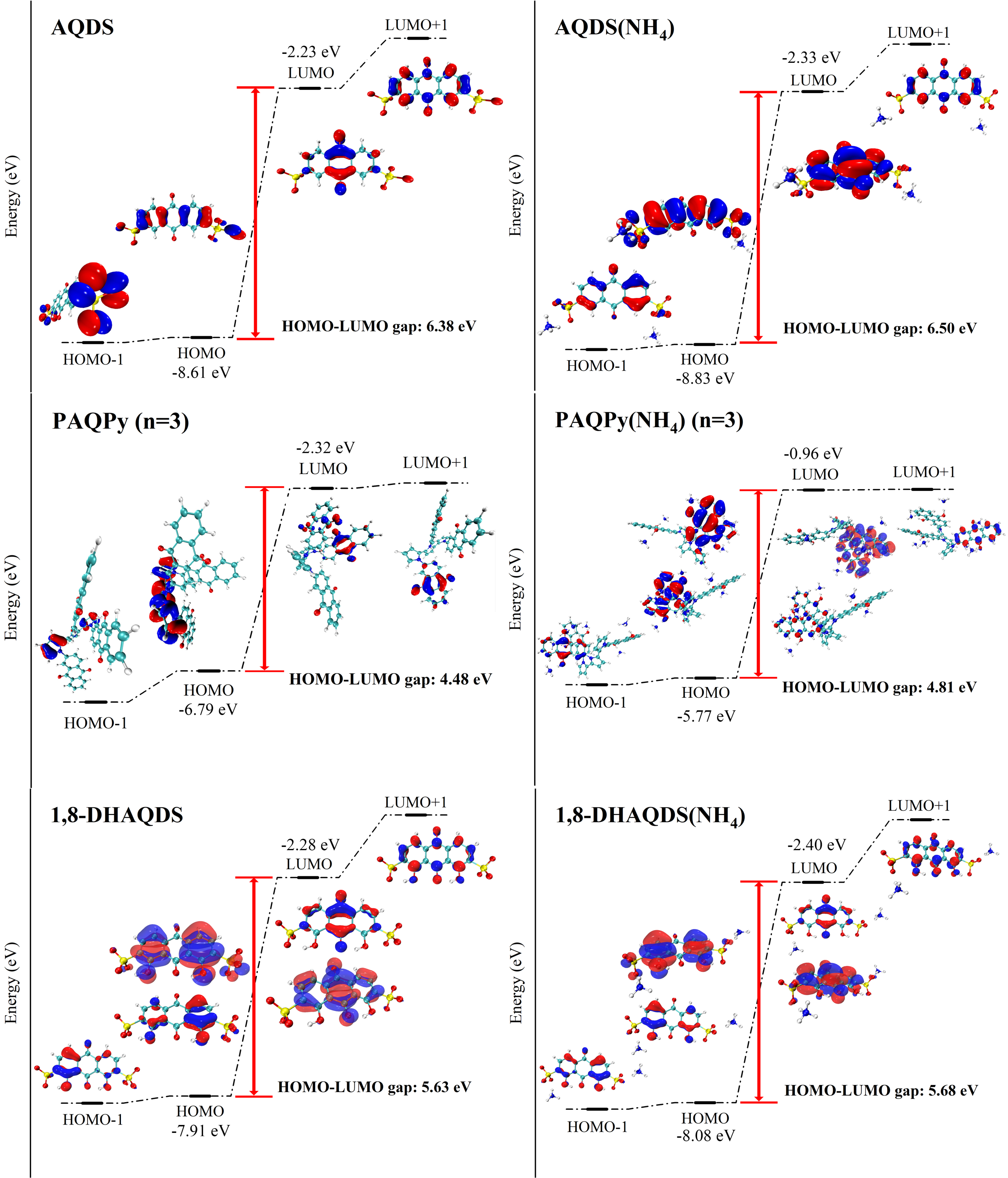
图 4. 含/不含 NH4+的 AQDS、PAQPy 和 1,8-DHAQDS 的 HOMO-LUMO gap。
要点四:通过DFT计算与实验方法验证预测方案的精确性
AQDS(NH4)和 DHAQDS(NH4)的 RP 都随着 pH 值的降低而升高,这与 DFT 计算关于有机化合物与 H3O+ 相互作用引起的 RP 移动的预测一致。氢键浓度与 H+ 和电解质浓度相关。根据电子传递动力学,这只影响电子传递速率和峰值电流,而 RP 保持不变。O-H...O 相互作用比 N-H...O 具有更强的键能,从而表明有机分子和 NH4+ 在溶剂酸度增加时形成的氢键之间存在明显的差异。NH4+ 比 pH 值调节提供了更精确的控制区间,可以在较窄的范围内精确、定量地调节分子 RP。这些发现强调了 N-H...O 氢键作为 NH4+ 和有机分子之间主要相互作用的重要作用,从而验证了 DFT 计算所确定的通过氢键调节 RP 的假设机制。与仅使用 1,8-DHAQDS 的 AORFB 相比,该 RT-AORFB 的放电容量从 562.62 mAhL-1 显著提高了 3.86 倍,达到 2169.23 mAhL-1。有了这些令人鼓舞的结果,预计这项工作中的 DFT 方案可以作为预测和设计RT-RFB 中 RM 和 SM 的有效方法,适用于大规模储能。
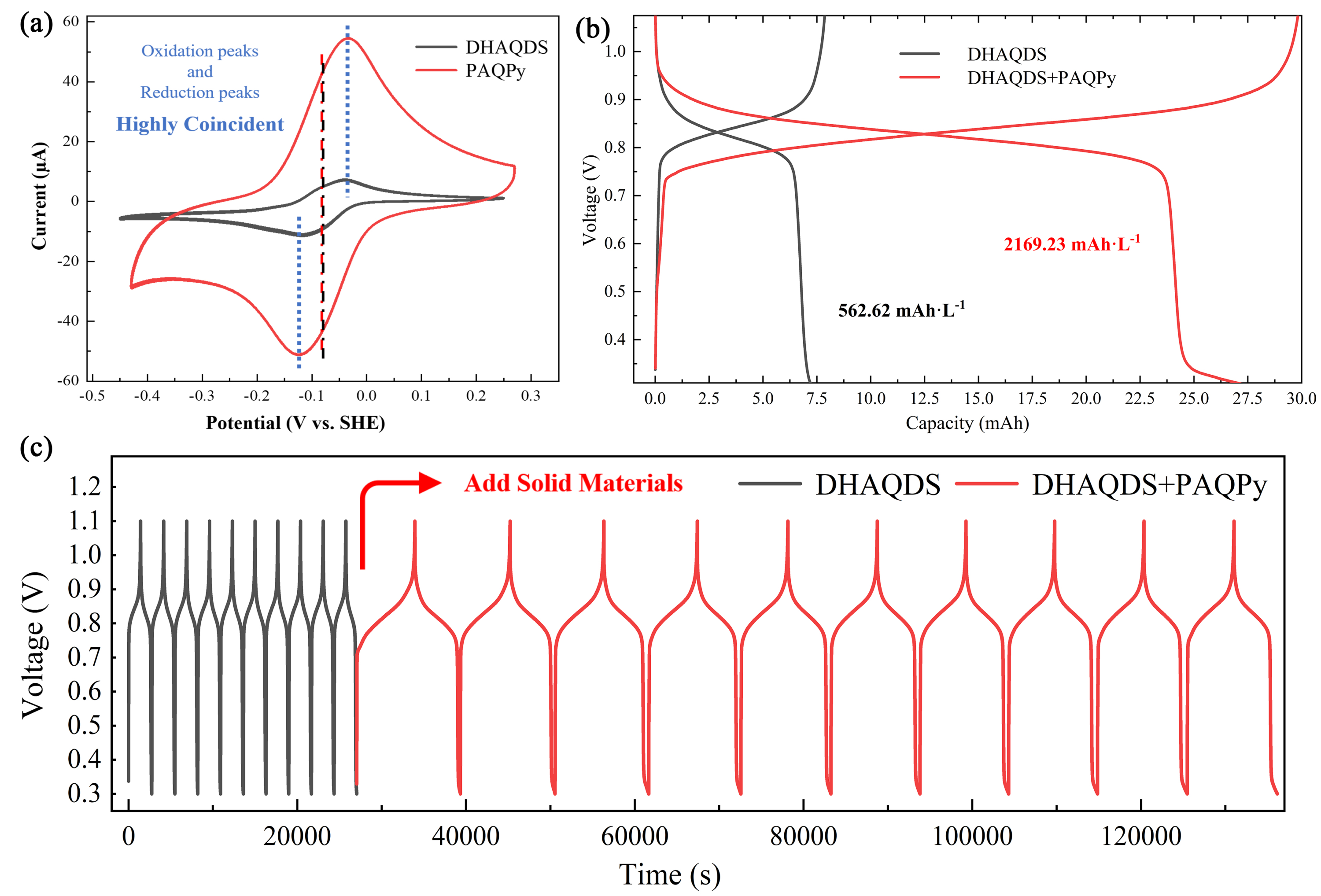
图 5. (a)12.86 mM 1,8-DHAQDS 和 PAQPy在 2.5 M H2SO4 中的 CV;在 13 mL 12.86 mM 1,8-DHAQDS 溶液槽中加入 0.3 g 固体前后 RT-AORFB 的电压曲线:(b)容量-电压曲线和(c)时间-电压曲线。
【原文链接】
https://doi.org/10.1016/j.ensm.2024.103389
【作者简介】

第一作者:荣思达,威廉希尔WilliamHill官方网站2022级博士研究生。研究方向:水系有机靶向液流电池。

通讯作者:纪亚,威廉希尔WilliamHill官方网站助理教授,博导,上海市领军人才计划获得者。分别从南京大学和新加坡国立大学获得学士学位和博士学位。研究方向为高能靶向液流电池和液流电池关键材料。曾在Advanced Functional Materials,Energy Storage Materials,Journal of Materials Chemistry A,Journal of Membrane Science等国际期刊上发表论文20余篇。担任Carbon Neutrality期刊青年编委,客座编辑。

 友情链接 ---
友情链接 ---